Ailesel lipoprotein Lipaz Eksikliği
Ailesel olarak nesilden nesile aktarılan bir lipoprotein lipaz eksikliği, bir kişinin yağ moleküllerini parçalamak için gerekli bir protein olan lipoprotein lipaz enziminden yoksun kaldığı, genetik bir hastalıktır.
HASTALIĞIN DİĞER İSİMLERİ
• Ailesel LPL eksikliği
• Hiperlipoproteinemi tip 1
• Burger ve Grutz sendromu
ÖZET
Ailesel olarak nesilden nesile aktarılan bir lipoprotein lipaz eksikliği, bir kişinin yağ moleküllerini parçalamak için gerekli bir protein olan lipoprotein lipaz enziminden yoksun kaldığı, genetik bir hastalıktır.
Bu enzimin eksikliği, etkilenen kişilerin belirli yağları sindirmelerini önler. Bu, kandaki şilomikron adı verilen yağ damlacıklarının birikmesine ve trigliseritlerin kan konsantrasyonunda bir artışa neden olur.
Belirtileri karın ağrısı, tekrarlayan pankreas iltihabı (pankreatit), karaciğer ve / veya dalağın anormal genişlemesi (hepatosplenomegali) ve erruptif ksantoma olarak bilinen cilt lezyonlarının gelişmesini içerir.
Ailesel lipoprotein lipaz eksikliği LPL genindeki değişikliklerden (mutasyonlar) kaynaklanır. Otozomal resesif paternde kalıtsaldır. Tedavi yöntemi olarak, semptomları ve kan trigliserit düzeylerini çok az yağlı bir diyet ile kontrol etmeyi amaçlayan bir yol izlenir. Bireysel semptomların tedavisi (yani pankreatit), belirlenmiş tedavi kılavuz ilkelerinin izlenmesini içerir.
TANIM
Chylomicronemia sendromu semptomlar için genel bir terimdir. Chylomicronemia sendromunun birçok nedeni vardır. Ailesel şilomikronemi terimi bazen lipolizin eş anlamlısı olarak kullanılır. Bununla birlikte, ailesel chylomicronemia'nın farklı nedenleri vardır. Geçmişte, ailesel lipoprotein lipaz eksikliği, 1932'de Drs tarafından hiperlipoproteinemi tipi olarak adlandırılmıştı. Burger ve Grutz sendromu olarak.
İşaretler ve Belirtiler
Ailevi LPL eksikliği vakalarının çoğu, çocukluk döneminde, genellikle 10 yaşından önce tanımlanır. Vakaların yaklaşık yüzde 25'inde, hastalık yaşamın ilk yılında belirlenir. Bazı vakalar yetişkinliğe kadar tanımlanamayabilir. Örneğin, bazı kadınlara hamile kaldıktan sonra veya kontraseptif ilaç almaya başladığında tanı konulabilir.
Ailesel LPL eksikliğinin ciddiyeti, bir bireyin diyetindeki yağ miktarına bağlı olarak değişen chylomicronemia derecesine bağlı olarak değişir. Başlıca belirtileri karın ağrısı, pankreatit, erüptif ksantomalar ve hepatosplenomegalidir.
Ailesel LPL eksikliğinin en sık semptomu epizodik karın ağrısıdır. Karın ağrısının şiddeti hafif ila şiddetli arasında değişebilir ve bazı durumlarda güçsüz olabilir. Ağrı, karnın üst, orta bölgesinde (epigastrik bölge) yer alabilir ve sırt ağrısına yol açacak şekilde yayılabilir. Bazı durumlarda, ağrı yaygın olabilir (dağınık) ve akut karına (peritonit) potansiyel olarak benzeyebilir. Geçmişte, bu doğru tanı koyulamadığı için gereksiz ameliyat neden oldu.
Ailesel LPL eksikliği olan kişilerde karın ağrısı, tekrarlayan pankreas iltihabı olaylarından (pankreatit) kaynaklanabilir. Pankreas mide arkasında bulunan küçük bir bezdir. Pankreas, sindirim ve vücutta uzmanlaşmış rolleri olan hormonlara yardımcı olmak için bağırsaklara giden enzimleri salgılar. Pankreatitin ana semptomu, bazen yoğun olan ve en sık karın bölgesinin sol üst veya ortasında hissedilen ağrıdır. Pankreatit ayrıca mide bulantısı, terleme, halsizlik, titreme, rutubetli cilt ve cildin veya gözlerin beyazlarının hafif sararmasına neden olabilir (sarılık). Bazı kişiler de potansiyel olarak öldürücü olabilecek akut, tekrarlayan pankreatit geliştirir.
Kronik pankreatit, diyabet, pankreasın kalsiyum tuzlarının birikmesinden dolayı pankreasın sertleşmesi (pankreas kalsifikasyonu) ve aşırı miktarda yağ içeren dışkıları köpüklü, kir kokulu ve yüzdürücü (steatorit) gibi ek komplikasyonlarla ilişkili olabilir. Bununla birlikte, bu komplikasyonlar ailesel LPL eksikliği olan kişilerde olağandışıdır. Tekrarlayan pankreatit atakları olan kişilerde bile, bu tür komplikasyonlar nadiren orta yaşlara kadar gelişir. Nadir olmasına rağmen, LPL eksikliğindeki pankreatit, hayatı tehdit eden ciddi komplikasyonlara neden olabilir.
Karaciğer ve dalağın büyümesi (hepatosplenomegali), özellikle bebeklerde ve küçük çocuklarda ortaya çıkabilir. Büyütme derecesi, genellikle diyetteki yağ miktarıyla birlikte değişir. Hepatosplenomegali, özel bir makrofaj türünün birikmesinden kaynaklanır. Makrofajlar yabancı veya zararlı maddeleri yutan beyaz kan hücreleridir. Ailesel LPL eksikliğinde makrofajlar fazla miktarda trigliserit alır ve köpük hücrelerine dönüşür. Köpük hücreleri, vücutta aşırı yağla başa çıkmaya çalışan ve genellikle yağ malzemeleri içeren özel makrofajlardır. Ailesel LPL eksikliği olan bireylerde köpük hücreleri anormal kemik iliği, karaciğer ve dalakta birikir.
Etkilenen bireylerin yaklaşık yüzde 50'si, belirli yağlardan (lipidler) oluşan cilt lezyonları olan erüptif deri ksantomaları geliştirir. Ksantomalar ciltte kabarık, kırmızımsı sarı renkli çarpma veya nodül şeklinde görünebilir. Genellikle kalçalarda, dizlerde ve dış kollarda görülürler.
Bireylerin lezyonları yaklaşık 1 milimetre büyüklüğünde ölçebilir, ancak ksantomalar sıklıkla kümelenir ve daha büyük lezyonlar oluşturmak için birlikte büyür (birleşebilir). Eruptif ksantomalar, tekrarlanan travma veya aşınmaya maruz kaldıkları bir bölgede gelişmezlerse, genellikle acı verici veya hassas değildirler.
Ksantomalar genellikle plazmada trigliserit seviyeleri artmaya başladıktan birkaç gün sonra ortaya çıkar. Yağlı, sarımsı bir madde ve bazen sütlü bir sıvı içerebilirler. Ksantomalar, plazmadaki trigliserit miktarı azaldıkça haftalar ila aylar arasında kaybolur. Ailesel LPL eksikliği olan bireylerde ksantomaların kalıcı varlığı, trigliserit düzeylerini düşürmek için yetersiz bir tedavi uygulandığını gösterir.
Bazı durumlarda, ailesel LPL eksikliği olan bireylerde, depresyon, hafıza kaybı ve demans dahil olmak üzere çeşitli geri dönüşümlü nöropsikiyatrik bulguları içeren ek semptomlar olabileceği gözlemlenmiştir.
Ailesel LPL eksikliği olan bazı kişiler, potansiyel olarak koroner kalp hastalığına veya periferik vasküler hastalığa neden olan, yağ materyali birikimine bağlı olarak çeşitli kan damarlarının kalınlaşması ve tıkanması ile karakterize erken ateroskleroz geliştirmiştir. Bununla birlikte, çoğu araştırmacı, ailesel LPL eksikliği olan bireylerin, ateroskleroz gelişme riskinde artış olduğuna inanmamaktadır.
OLUŞUMUNA NEDEN OLDUĞU DÜŞÜNÜLEN ETKENLER
Ailesel LPL eksikliğine LPL geninin mutasyonları neden olur. Bu genetik mutasyon, otozomal resesif bir özellik olarak kalıtsaldır. Genetik hastalıklar, baba ve anneden alınan kromozomlar üzerindeki belirli bir özellik için genlerin kombinasyonu ile belirlenir.
Resesif genetik bozukluklar, bir birey her bir ebeveynden aynı özellik için aynı anormal geni aldığında ortaya çıkar. Bir birey hastalık için bir normal gen ve bir gen alırsa, kişi hastalık için taşıyıcı olacaktır, ancak genellikle semptomlar kendini göstermez. İki taşıyıcı ebeveynin hem kusurlu geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte% 25'tir. Ebeveynler gibi taşıyıcı olan bir çocuğa sahip olma riski her hamilelikte% 50'dir. Bir çocuğun her iki ebeveynden normal gen alma ve bu özellik için genetik olarak normal olma şansı% 25'tir. Risk erkeklerde ve kadınlarda aynıdır.
LPL GENİ
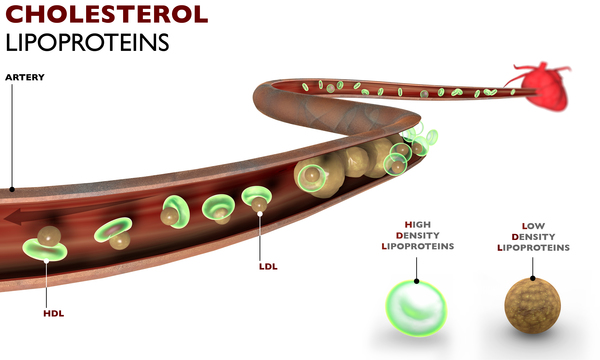
LPL geni, lipoprotein lipaz olarak bilinen bir enzimi oluşturmak (kodlamak) için talimatlar içerir. Bu enzim vücuttaki bazı yağların uygun şekilde parçalanması için esastır. Yağ diyet yoluyla elde edilir ve bağırsaklar tarafından emilir. Şilomikronlar olarak bilinen büyük lipoproteinler tarafından trigliserit şeklinde taşınır. (Trigliseritler, vücut hücreleri tarafından yakıt için kullanılan yağ molekülleridir.) Şilomikronlar kan dolaşımına salındığında, apolipoprotein C2 adı verilen şilomikronlar içindeki bir protein aktive edilir. Bu protein, lipoprotein lipaz enzimi tarafından tanınır ve sonuçta trigliseritin parçalanmasına neden olur. Lipoprotein lipaz yetersiz veya yetersiz olduğunda, plazmada chylomicrons birikir ve bu da sırayla plazmada anormal miktarda trigliserit birikmesine neden olur. Kandaki fazla trigliserit ve şilomikronların birikmesi, ailesel LPL eksikliğinin belirtilerine neden olur.
Individuals who inherit one mutated gene and one normal gene (heterozygotes) do not develop familial LPL deficiency. However, these individuals may have a slightly increased risk of developing mixed dyslipidemia with low HDL cholesterol levels. Heterozygotes may also be more susceptible to developing atherosclerosis than non-carriers, especially if they gain weight or remain on a high fat diet.
Teşhis
Ailesel LPL eksikliğinin teşhisi, karakteristik semptomların tanımlanması, ayrıntılı bir hasta öyküsü, ayrıntılı bir klinik değerlendirme ve kan testleri de dahil olmak üzere bazı testlere dayanarak şüphelenilebilir.
Klinik Test ve Çalışma
Kan testleri, heparinin intravenöz olarak uygulanmasının ardından, plazmada lipoprotein lipaz enziminin azalmış aktivitesini ortaya çıkarabilir. Heparin normalde karaciğerde bulunan ve vücutta lipoprotein lipaz salınımını uyaran bir maddedir. Ailesel LPL eksikliğinin teşhisi, LPL genindeki mutasyonlar için moleküler genetik test ile doğrulanabilir. Moleküler genetik testler, ticari ve akademik araştırma laboratuvarları aracılığıyla yapılabilir.
GÖRÜLME SIKLIĞI
Ailesel LPL eksikliği erkekleri ve kadınları eşit sayıda etkiler. Son verilere dayanarak (2013), genel popülasyondaki yaklaşık 250.000 kişiden 1'inde gerçekleştiği tahmin edilmektedir ve tüm yarışlarda tanımlanmıştır. Kurucu etkisinden dolayı, Quebec, Kanada'da yaygınlık çok daha yüksektir. Bir kurucu etki, küçük bir izole edilmiş yerleşimci popülasyonunun (kurucuların), belirli bir genetik özelliğin yüksek prevalansına yol açan birkaç kuşak boyunca genişlemesidir.
Çeviren ve Derleyen: Caner ERDOĞAN
Kaynaklar:
https://rarediseases.org/rare-diseases/familial-lipoprotein-lipase-deficiency/
https://rarediseases.info.nih.gov/diseases/12241/familial-lipoprotein-lipase-deficiency
Genetik Haberleri
-

Allopatrik türleşme nedir ? Nasıl Gelişir ?
-
Maryland’teki “Kölelerin” Yaşayan 42.000 Akrabası Bulundu
-
Araştırmacılar kediler, yunuslar, kuşlar ve düzinelerce başka hayvanın genom haritasını çıkarıyor
-
Kolombiya'da nadir görülen bir kuş türünde "gynandromorphy" gözlemlendi
-
Kurumaya dayanıklı bitkiler için genom veritabanı yayınlandı
-
En son DNA barkodlama teknolojisiyle İsrail'in tatlı su balık türleri listesinin yeniden gözden geçirilmesi
-
İnsanların Daha Önce Bilinmeyen Bir Dokunma Duyusu Keşfedildi
-
Bilim İnsanları Tüm İnsan Genomunun Dizilimini Çıkardı. Ancak Henüz Bitmedi
-
İlk Defa Tazmanya Kaplanından RNA Elde Edildi
-
Neandertal DNA’nız, Sizi Acıya Karşı Daha Hassas Yapıyor Olabilir
-
Epigenetik ve Epigenetik Mekanizmalar
-
İlk taslaktan 20 yıl sonra insan Y kromozomu tamamen dizilendi.
-
Kim Bu Kimerizm? Tek Bedende İki Kişi
-
Gen terapi, genetik materyalin yeniden düzenlenmesi
-
mRNA Aşıları: Genetik İnovasyonunun Yeni Yüzü ve Sağlıkta Devrimi
